 Alzheimer’s disease (AD), the most prevalent form of dementia in the elderly, presents with a complex etiology that hinders the development of effective therapies; leaving unmet needs for the more than 6 million Americans diagnosed with this devastating disorder. Symptoms of AD primarily manifests as progressive memory loss and declining cognitive abilities. Beyond altered proteostasis and accumulation of amyloid-beta and Tau in the brain, disruption of circadian rhythms and epigenetic deregulation are emerging as important contributors to AD pathogenesis.
Alzheimer’s disease (AD), the most prevalent form of dementia in the elderly, presents with a complex etiology that hinders the development of effective therapies; leaving unmet needs for the more than 6 million Americans diagnosed with this devastating disorder. Symptoms of AD primarily manifests as progressive memory loss and declining cognitive abilities. Beyond altered proteostasis and accumulation of amyloid-beta and Tau in the brain, disruption of circadian rhythms and epigenetic deregulation are emerging as important contributors to AD pathogenesis.
Circadian rhythms are generated by the oscillation of clock genes in transcription/translation feedback loops and epigenetic mechanisms are intimately involved in regulating this process. Behavioral circadian alterations, collectively known as sundowning, are experienced by more than 80% of patients and represent leading factors for hospitalization and nursing home placement. Importantly, emerging data strongly support a causal role for circadian disruption in AD pathophysiology, including exacerbation of inflammation and amyloid-beta accumulation, cognitive impairment, and oxidative stress.
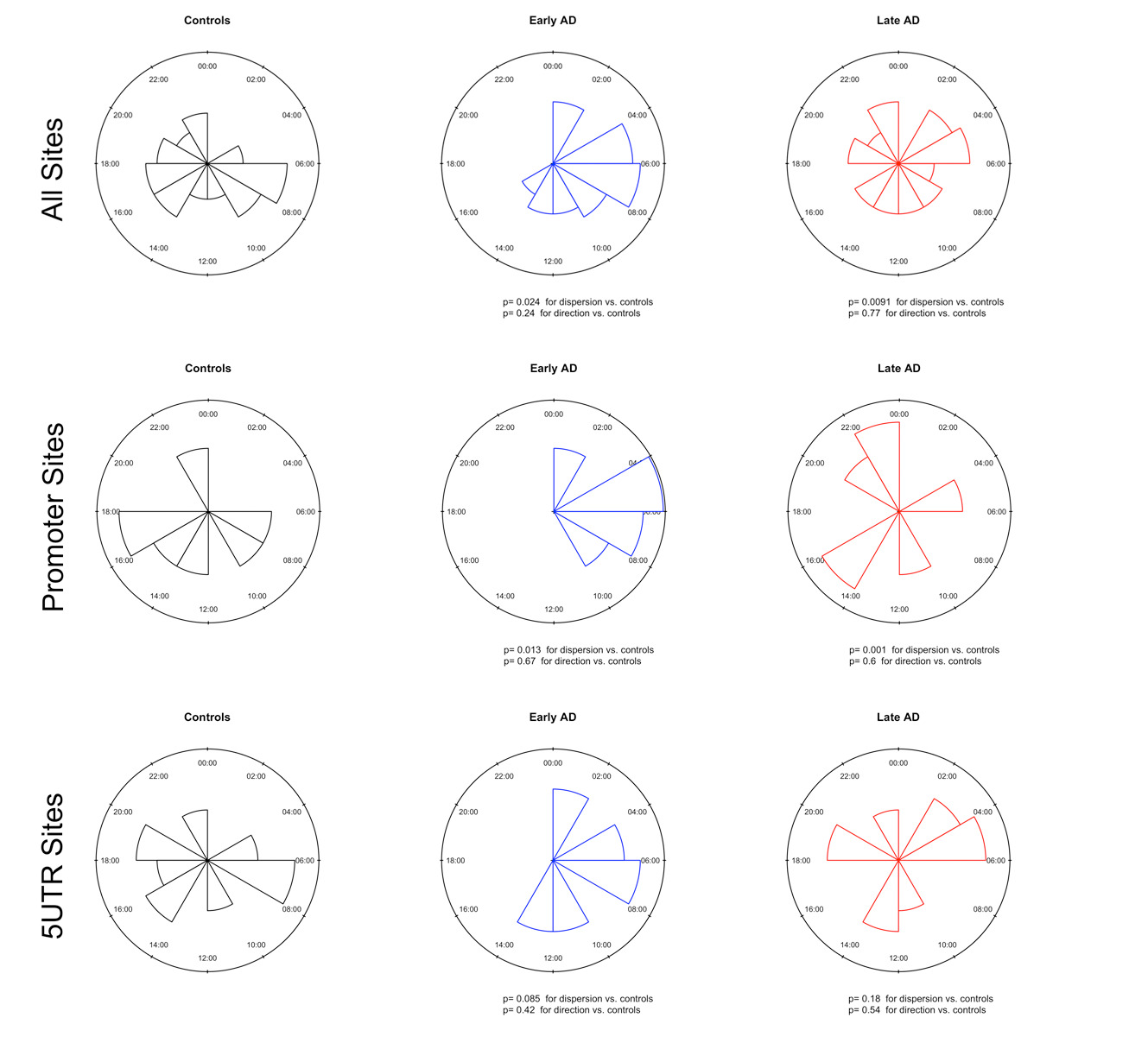
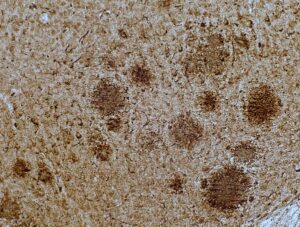
Circadian factors may regulate gene expression by modulating epigenetic mechanisms, and while both are altered in the brains of patients with AD, they are poorly characterized. By profiling genome-wide methylation in more than 70 postmortem midfrontal cortex samples from the UCSD Shiley-Marcos Alzheimer’s Disease Research Center (ADRC), including early AD cases, advanced dementia patients, and healthy control subjects, we uncovered alterations in DNA methylation of several circadian-controlled genes. Further investigation focused on BMAL1, the positive regulator of the molecular clock, and identified multiple genomic sites that present rhythmic methylation levels and which were altered in early AD. This epigenetic deregulation impacts rhythmic expression of BMAL1 mRNA and protein levels in the brain.
Our current goal is to define the pathways deregulated by circadian clock dysfunction that contribute to neurodegeneration in AD, with particular focus on epigenetic mechanisms.
Our studies are set to provide a comprehensive characterization of circadian regulation in AD at an unprecedent resolution through the use of cutting-edge multi-omics technologies, behavioral characterization, metabolic analysis, as well as investigation of sleep architecture and neuropathology. The results of these experiments will provide a baseline for future studies in the field. DNA methylation-seq, ATAC-seq, and RNA-seq analyses are currently undergoing, and we expect to delineate the circadian organization of the chromatin and transcriptomic landscapes of different brain regions in the APP23 transgenic mouse model of AD. In addition, we are pioneering the application of recently developed spatial transcriptomic platforms to generate temporal, spatial, and cell-specific pathways of gene expression in the brain.
Capitalizing on our findings, we are already testing different interventions to modulate the molecular clock as a strategy to slow disease progression.